
ATAC下游分析

ATAC下游分析
kuteliyafukaatac下游分析整理
得到所有样本的.Narrowpeak文件后,依次进行peak merge/insertion count,即可得到所有样本peak的矩阵。
行为各个peak区域的起始位点在染色体上的坐标,列是各个样本。
注意:下面两个输入路径都是nf-core中atac pipeline的输出结果
peak merge
script:
1 | Rscript ./createIterativeOverlapPeakSet.R \ |
需要准备:.R文件+hg.blacklist.bed+file.txt。文件内容如下:
数据表
R文件下载
hg黑名单
得到All_Samples.fwp.filter.non_overlapping.bed
insertion count
之后进入从mj那里拿到的aging环境。
运行insertion_count.R脚本,修改脚本最上方的三个路径。
脚本如下:
insertion_count.R脚本
得到count_matrix.csv
peak clustering
合并重复样本,对样本进行重命名+排序。
这里尝试过多种方法,z-score/wilcoxon检验/k-means clustering。经过比较k-means clustering最为合理。
代码如下:
kmeans.ipynb脚本
view_result脚本
对所有peak进行clustering,k=5。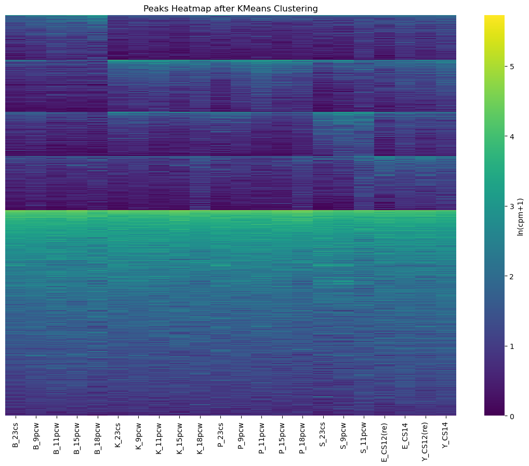
这些peaks与组织基本一致,之后再取出每个组织,再对其进行k-means聚类,得到temporal-dynamic peaks。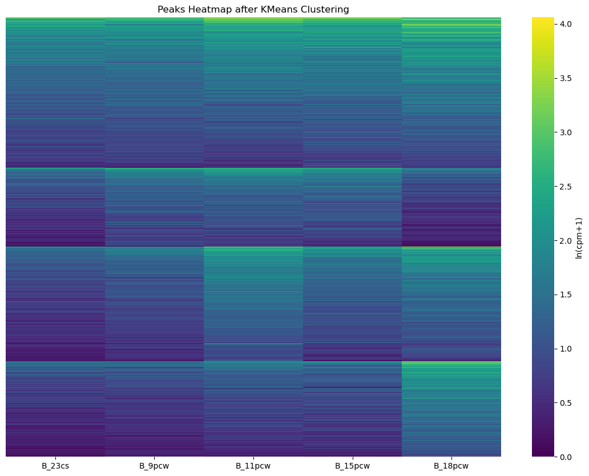
得到各个组织的差异.bed文件,以及所有peak地.bed文件。
以所有peak为背景,对各个组织的差异peak依次通过great进行go富集以及motif富集(kai的snapatac2)
great富集
在great网页(https://great.stanford.edu)
直接在网页中使用(似乎R语言中的已经停用了,没法在代码中用)
motif富集
这里首先要从meme suite(https://meme-suite.org/meme/db/motifs) 中下载人类的转录因子.meme 。
接着通过代码通过snapatac2进行motif富集,代码如下
motif_enrichment
再通过代码view_result去检查/绘图。
转录因子筛选
结合要来的单细胞数据,可以粗略地分析
有三个条件:
- gene expression高
- atac数据能够enrich motif
- 相比其它样本更高
分析步骤:
以brain样本为例。首先,对于所有样本的peak,由k means得到若干peak region的.bed文件。
对于这些.bed文件,进行snapatac2的motif_enrichment富集,得到若干富集结果。代码
对于富集结果进行分析,选择top100的motif,保存。代码
读取scRNA数据,检查这100个motif对应的转录因子在rna上的表达量。 代码



