
structure_study1

structure_study1
kuteliyafuka如何通过alphafold3和pymol研究蛋白质与核酸与原子互做
Alphafold3使用
首先,在Alphafold3的官网上,找到example3:Protein-DNA-ion: PDB 7RCE。
预测结构之后,下载,找到0.cif文件,用pymol打开。
Alphafold3中个参数含义:
模型颜色
局部折叠的置信度pLDDT(predicted local distance difference test) 衡量的是AF3对每一个残积的自信程度。
注意:低置信度的区域很少是因为AF3算错了,往往是因为这段序列在现实中没有固定形状,通常出现在蛋白质的末端或者天然无序区域(IDPs)。
全局得分
pTM(predicted template modeling score),范围是0-1,>0.8意味着高质量模型。
ipTM(interface pTM),专门评估结合界面准确度的分数,>0.8,说明AF3对预测的相互作用有信心
PAE(predicted aligned error),一张颜色矩阵,颜色越深代表越可靠(每个残基和每个残基之间的相对位置吗?)。
pymol可视化
将cif拖入pymol结果如下: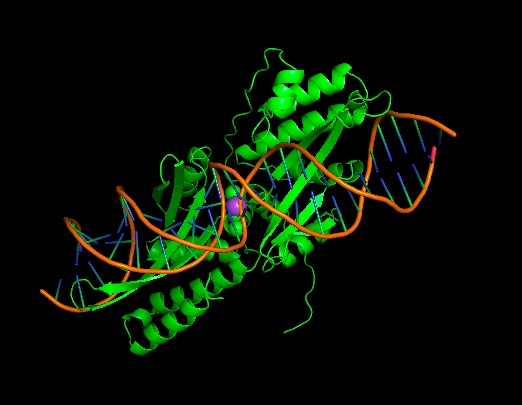
目的:找到与离子互做的氨基酸。
第一步:大扫除与宏观上色
刚导入的模型往往会把所有原子都画成线条,看起来像一团乱麻。我们先把它清理干净,并区分出蛋白质和 DNA。
1 | # 1. 隐藏所有默认的杂乱线条 |
此时,屏幕上应该只剩下灰色的蛋白质束和橙色的 DNA 双螺旋了。
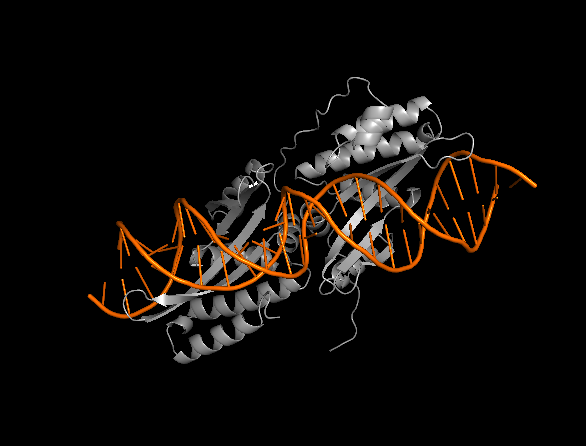
第二步:揪出那个“小透明”离子
1 | # 1. 创建一个名为 "my_ion" 的选择集,包含常见的金属离子 |
现在能在灰色和橙色的交界处(或蛋白质的某个深坑里),看到一个或几个闪闪发光的黄色小球。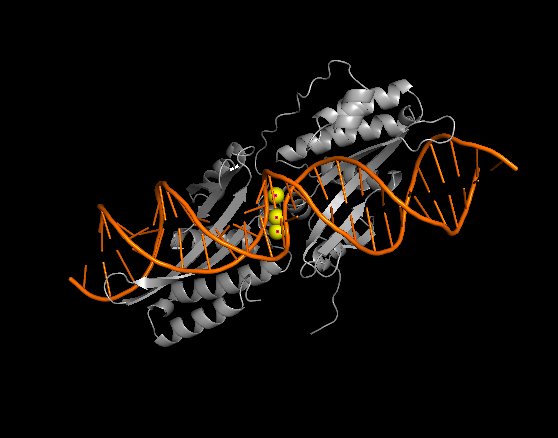
寻找“幕后黑手”(抓取离子的氨基酸)
我们要让软件自动找出距离这个黄色小球 4.0 埃 (Å) 以内的所有氨基酸和核苷酸,它们就是抓住离子的“爪子”。
1 | # 1. 选中距离离子 4.0 埃以内的所有完整残基,并命名为 "claws" (爪子) |
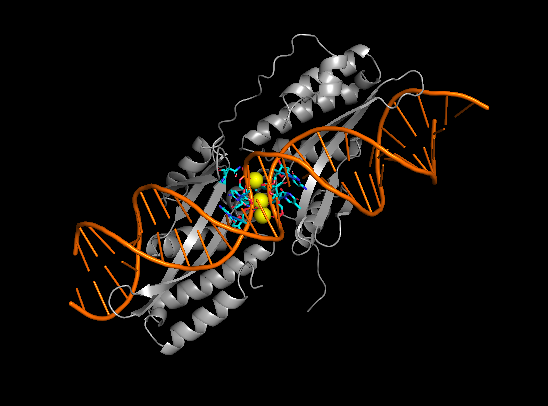
此刻,能够清晰地看到几个氨基酸(比如带有环状结构的组氨酸,或者带硫原子的半胱氨酸)像夹娃娃机的爪子一样,从四面八方精准地指向那个黄色的小球。
画出相互作用力
为了让图表具有绝对的科学说服力,我们需要把离子和“爪子”之间的相互作用力(配位键或静电作用)用虚线画出来。
1 | # 测量离子与爪子之间距离在 3.5 埃以内的原子,并用虚线连接,命名为 "my_bonds" |
标记氨基酸
在复杂的蛋白质结构里,如果让所有原子都显示标签,屏幕瞬间就会变成一团看不清的“乱码”。所以,结构生物学家的常规操作是:只在氨基酸的“骨架节点”(α 碳原子,简写为 CA)上打标签。
1 | # 1. 提取氨基酸的名字 (resn) 和编号 (resi),并只贴在 CA 原子上方 |
能够看到五个aa,GLN 203,GLU 21,GLY176,ALA20,ASP177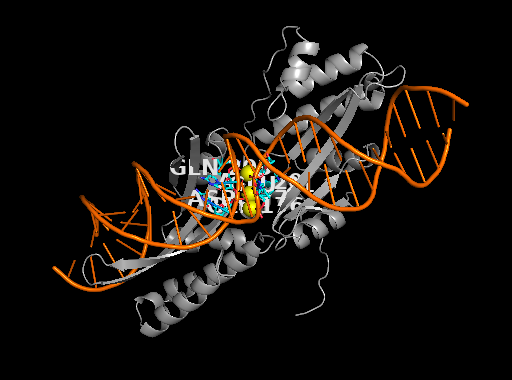
视觉优化
可视化遇上以下三个问题:
镜头太远:核心作用区域只占了屏幕很小的一部分。
标签打架:字号(22)对于密集的原子来说太大了,全挤成了一团白色的乱码。
背景喧宾夺主:巨大的灰色蛋白质和橙色 DNA 挡住了内部精细的化学键细节。
改进:
- 镜头拉进和背景虚化
让我们的视线穿过厚重的蛋白质,直接聚焦在离子身上。
1 | # 1. 镜头拉近:精准聚焦到离子周围 8 埃的微观世界 |
- 抢救堆叠的标签
我们要把字号改小,并且把标签往原子的右上方“推”开一点,防止它们互相重叠。
1 | # 1. 把字号改小到 14 |
- 加粗爪子与渲染
最后,我们把细节加粗,换上干净的白底,并开启光影追踪渲染。
1 | # 1. 让结合的“爪子”和“虚线”变得更粗壮、更有力量感 |
光影渲染和导出
1 | # 1. 把背景换成干净的白色 (论文排版必备) |
现在,屏幕上有了蛋白、DNA、发光的离子、配位键的虚线以及标签。但默认的黑色背景和塑料质感看起来还不够高级。按以上代码运行得到了高清白底插图。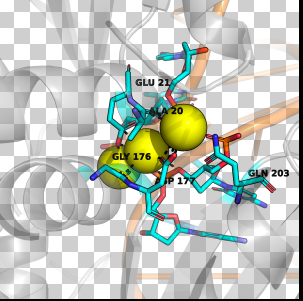
结构叠合
我们可以直接在 PyMOL 里用一句命令从 PDB 数据库下载真实的 7RCE 实验结构,然后把它和我们 AF3 预测的模型叠在一起,看看 AI 算出来的 DNA 螺旋角度、钙离子的位置,和现实世界中做实验拍出来的究竟有多大误差!
- 从PDB下载蛋白质结构
PyMOL 内置了直接连接全球蛋白质数据库(PDB)的接口。
1 | # 直接从 PDB 数据库下载真实的 7RCE 实验结构 |
屏幕上应该出现了两套重影的复合物,它们可能不在同一个位置,甚至互相穿模。
2. 给真实结构上色
为了不和刚才 AI 预测的灰色蛋白、橙色 DNA 混淆,我们给“真实版”换一套显眼的冷色调。
1 | # 给真实的蛋白质穿上“淡青色”外衣 |
- 叠合模型
这是结构生物学里最经典的一行命令。它会让电脑计算两套坐标,然后像套娃一样把它们强行重合在一起。
1 | # 把 AI 预测的模型,叠合(align)到真实的 7rce 模型上 |
此时能够看到pymol上方控制台出现的一行小字: Executive: RMSD = 0.622 (2451 to 2451 atoms)
RMSD(均方根偏差)是评价预测准不准的黄金标准,衡量的是两套原子的平均空间距离差异。
RMSD < 2.0 埃:极其优秀,AI 几乎完美复现了现实。
RMSD < 1.0 埃:神级预测,AI 的精度已经达到了实验误差的极限(相当于“满分”)。
RMSD > 4.0 埃:预测失败,折叠完全不对。
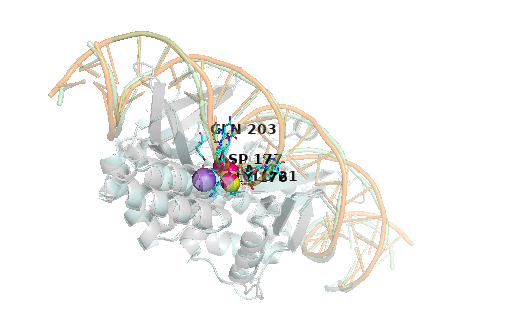
放大仔细看细节
看完宏观分数,我们来看看微观细节。实验结构里,那几个抓着钙离子的“爪子”(侧链)真的长那样吗?
1 | # 把镜头推近到真实的钙离子(亮粉色小球)周围 |
看宏观骨架: 灰色的 AI 蛋白和淡青色的真实蛋白是不是严丝合缝地套在一起了?橙色的 AI DNA 和淡绿色的真实 DNA 的螺旋角度有没有偏差?
看离子的位置(最震撼的一幕): 您会看到刚才黄色的 AI 钙离子和亮粉色的真实钙离子。它们是不是几乎完全重叠在了一起,像一个双色球?这意味着 AI 在没有任何实验指导的情况下,纯靠物理和进化规律,在庞大的空间里“盲猜”中了这个原子的绝对坐标!
看爪子的姿态: 观察两套氨基酸侧链(树枝状结构)。它们是不是像双胞胎一样指向了相同的角度?


